{kind=link}
Improved Five-Parameter Exponential-Type Potential Energy Model for Diatomic Molecules
Cite this Article
Fu Ke-Xue, Wang Meng, Jia Chun-Sheng. Improved Five-Parameter Exponential-Type Potential Energy Model for Diatomic Molecules. Communications in Theoretical Physics, 2019, 71(1): 103 
Permissions
Improved Five-Parameter Exponential-Type Potential Energy Model for Diatomic Molecules
† Corresponding author. E-mail:
Abstract
Abstract
The dissociation energy and equilibrium bond length as explicit parameters are used to establish an improved five-parameter exponential-type potential energy model for diatomic molecules. We demonstrate that the five-parameter exponential-type potential is identical to the Tietz potential for diatomic molecules. It is observed that the improved five-parameter exponential-type potential can well model the internuclear interaction potential energy curve for the ground electronic state of the carbon monoxide molecule by the utilization of the experimental values of three molecular constants.
Keyword:interaction potential energy;improved five-parameter exponential-type potential;carbon monoxide
1 Introduction
The knowledge of internuclear interaction potential energy between two atoms as a function of their relative positions is of fundamental importance in a wide variety of fields. There has been a growing interest in obtaining an available closed-form representation governing the interaction of two atoms for diatomic molecules in chemistry and physics. Numerous contributions have been made to build analytical internuclear potential energy functions.[1–15] In recent years, by utilizing the dissociation energy and equilibrium bond length for a diatomic molecule as explicit parameters, some authors[16–21] constructed improved versions with more convenient applications for some empirical potential energy functions, including the well-known Rosen-Morse, Tietz, Frost-Musulin, Manning-Rosen, and Pöschl-Teller potentials. The improved molecular potentials have received considerable attentions due to their real applications in many fields, including simulations of molecular potential curves, predictions of thermochemical quantities of gaseous substances, and calculations of molecular vibrational energies.[21–50] There has been an increasing recognition of the importance of the Tietz potential as a typical potential energy model. Hassanabadi et al.[51–53] studied the solutions of the Dirac equation and Klein-Gordon equation with the Tietz potential in terms of the supersymmetry quantum mechanics approach. Ikot et al.[54] investigated the bound state solutions of the Duffin-Kemmer-Petiau equation with the Tietz potential by employing the Nikiforov-Uvarov method. Onate et al.[55] computed the Shannon entropy and information energy under the modified Tietz-Hua potential. Khordad and Mirhosseini[56] studied the optical properties of spherical quantum dots by using the Tietz potential.
In 2001, the authors in the literature[12] proposed a five-parameter exponential-type potential model, and obtained a general energy spectrum formula by solving the Schrödinger equation with that in terms of the supersymmetry shape invariance approach. By choosing suitable forms of the parameters, the five-parameter exponential-type potential can turn to some well-known potential functions, including the Hulthén potential, Eckart potential, and improved Manning-Rosen potential. It is unfortunate that the model parameters in the five-parameter exponential-type potential lack explicit physical definitions. This fault has greatly affected its effective applications. In this work, we attempt to establish an improved five-parameter exponential-type potential energy model for diatomic molecules, and explore the relationship between it and the well-known Tietz potential in the presence of a diatomic molecule. We also apply the improved five-parameter exponential-type potential to model the internuclear interaction potential curve for the ground electronic state of the carbon monoxide (CO) molecule. A great deal of CO is released from tail gases, including coke oven gas, carbon black manufacturing tail gas, etc. CO is one of the most prominent gaseous environment pollutants, and a highly toxic gas for humans as well as for animals. Due to its high harmful effect on human health, the adsorption processes of CO in various adsorbent materials have aroused considerable attention.[57–61]
2 Improved Five-Parameter Exponential-Type Potential Energy Model
The five-parameter exponential-type potential energy model can be represented as Eq. (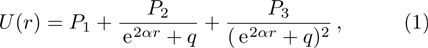
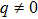
For a diatomic molecular potential energy function U(r), it is directly related to the three molecular constants and satisfies the following three relationships:

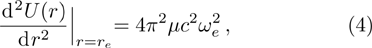
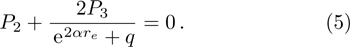
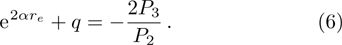

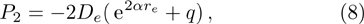
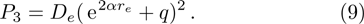
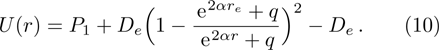
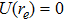
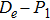
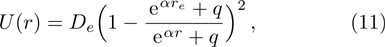
We calculate the second order derivation of U(r) given in expression (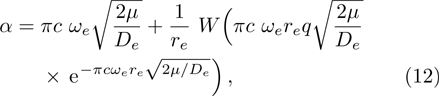
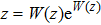
3 Applications
By choosing an available value of parameter q and putting the experimental values of the molecular constants De, re, and ωe as inputs, we reproduce the potential energy curve for the ground electronic state of CO in terms of the improved five-parameter exponential-type potential. The experimental values of three molecular constants are obtained by referring to the literature:[13] 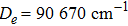
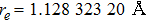
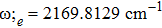
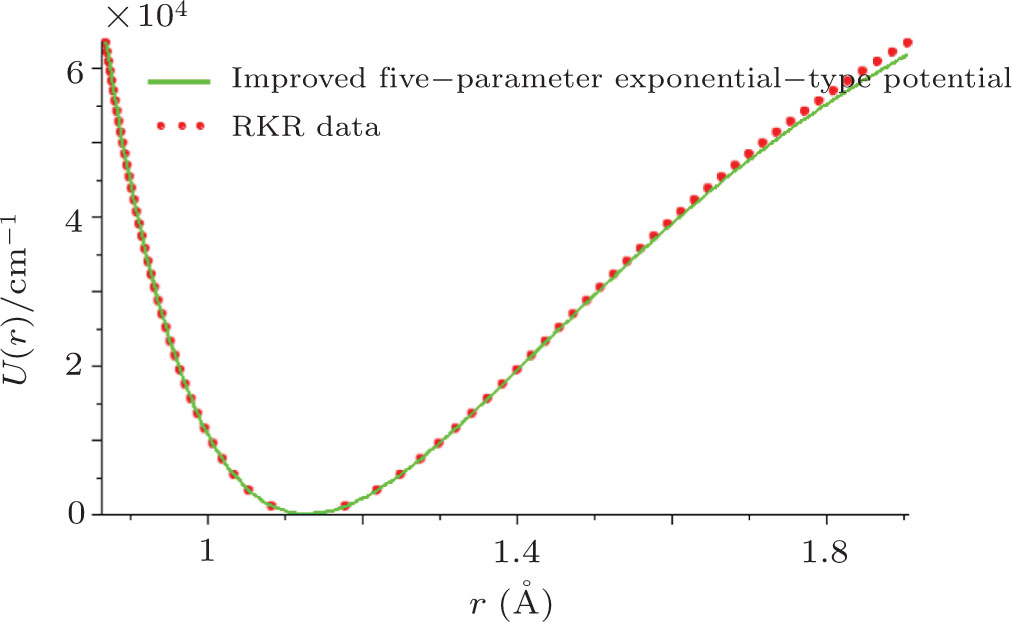
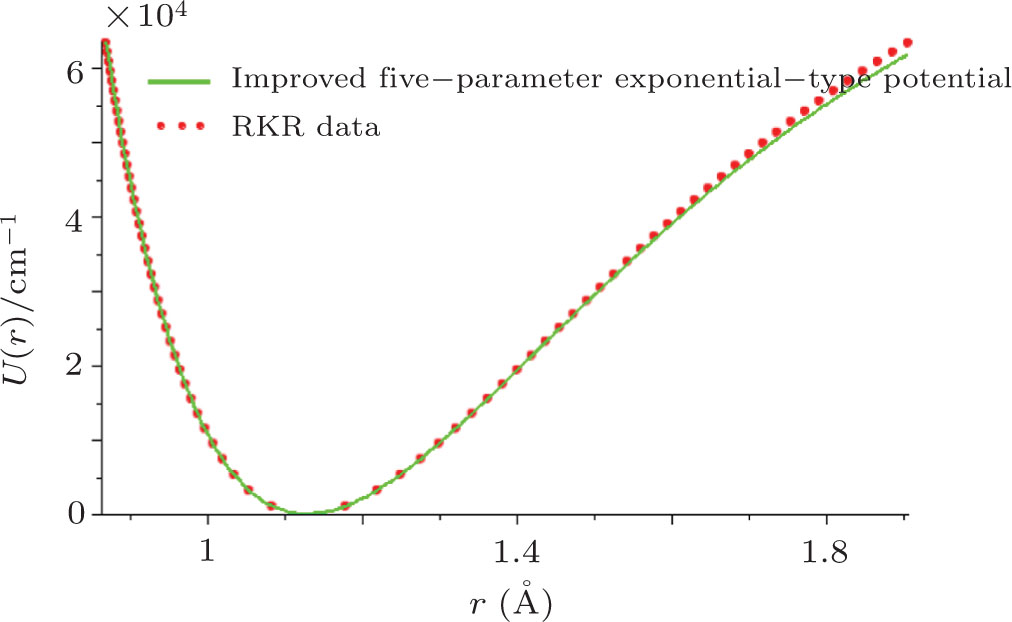 | Fig. 1. Experimental RKR data points and the improved five-parameter exponential-type potential energy curve for the ground electronic state of CO. |
In order to evaluate quantificationally the accuracy of the proposed potential, we consider the average absolute deviation of the improved five-parameter exponential-type potential versus the experimental RKR potential. The average absolute deviation is represented as 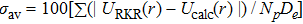
4 Conclusion
Through the introduction of the dissociation energy and equilibrium bond length as explicit parameters, we establish the improved five-parameter exponential-type potential model for diatomic molecules. We show exactly and simply that the five-parameter exponential-type potential is identical to the Tietz potential in the presence of a diatomic molecule. Using the experimental values of the dissociation energy, equilibrium bond length and equilibrium harmonic vibrational frequency, we model satisfactorily the potential energy curve for the ground electronic state of CO. By the calculation of average absolute deviation of the proposed potential versus the experimental RKR potential, we show quantitatively that the improved five-parameter exponential-type potential is an available model to represent the internuclear interaction potential for the ground electronic state of the CO molecule.
Acknowledgements
We would like to thank the kind referee for positive and invaluable suggestions which have greatly improved the manuscript.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] | |
| [57] | |
| [58] | |
| [59] | |
| [60] | |
| [61] | |
| [62] | |
| [63] | |
| [64] | |
| [65] | |
| [66] |