
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A First-Principles Study on the Vibrational and Electronic Properties of Zr-C MXenes
Cite this Article
Wang Chang-Ying, Guo Yong-Liang, Zhao Yuan-Yuan, Zeng Guang-Li, Zhang Wei, Ren Cui-Lan, Han Han, Huai Ping. A First-Principles Study on the Vibrational and Electronic Properties of Zr-C MXenes
. Communications in Theoretical Physics, 2018, 69(3): 336 
Permissions
A First-Principles Study on the Vibrational and Electronic Properties of Zr-C MXenes
† Corresponding author. E-mail:
Supported by the National Natural Science Foundation of China under Grant Nos. 11605273, 21571185, U1404111, 11504089, 21501189, 21676291, the Shanghai Municipal Science and Technology Commission 16ZR1443100, the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA02040104)
Abstract
Abstract
Within the framework of density functional theory calculations, the structural, vibrational, and electronic properties of ZrnCn − 1 (n = 2, 3, and 4) and their functionalized MXenes have been investigated. We find that the most stable configurations for Zr-C MXene are the ones that the terminal groups F, O, and OH locate on the common hollow site of the superficial Zr layer and its adjacent C layer. F and OH-terminated Zr3C2 and Zr4C3 have small imaginary acoustic phonon branches around Γ point while the others have no negative phonon modes. The pristine MXenes (Zr2C, Zr3C2 and Zr4C3) are all metallic with large DOS contributed by the Zr atom at the Fermi energy. When functionalized by F, O and OH, new hybridization states appear and the DOS at the Fermi level are reduced. Moreover, we find that their metallic characteristic increases with an increase in n. For (ZrnCn − 1)O2, Zr2CO2 is a semiconductor, Zr3C2O2 is a semimetal, and Zr4C3O2 becomes a metal.
1 Introduction
Recently, a great achievement has been made in the synthesization of new Zr-C materials.[1] The new produced Zr3C2Tz (T = F, O, OH) is composed by the Zr3C2 block and the functionalized group. It belongs to a new class of two-dimensional (2D) materials, MXene (Mn + 1XnTx), which is a type of 2D transitional metal carbide, nitride and carbonitride and can be prepared from layered nanolaminates by selective etching method.[1–2] The terminal groups T in MXene are usually oxygen (O), hydroxyl (OH) or fluorine (F). The currently researching hotspot of material science, MAX phase, is one kind of layered nanolaminates.[3–5] Taking Zr-C MXene as an example, its precursors can be Zrn + 1AlCn (n = 1, 2) (MAX phase), (ZrC)nAl3C2 (n = 2, 3, 4) and (ZrC)nAl4C3 (n = 2, 3) layered nanolaminates.[6]
In the past decades, more and more 2D materials are being investigated and synthesized for their excellent electronic properties, mechanical properties, optical properties and so on.[7–9] As the new discovered 2D materials, MXenes are currently expected to have numerous potential applications such as electronic devices[10–12] and electrode of Li ion batteries.[13–14] As the relatively early produced MXenes, 2D Ti-C and Mo-C materials have been explored by many works within the framework of density functional theory (DFT). However the knowledge about Zr-C MXenes is scarce due to the late preparation of 2D Zr-based materials. Considering the fact that DFT has gained substantial achievements in material field, a first-principles investigations on the 2D ZrnCn − 1 (n = 2, 3, 4) and their functionalized MXenes have been done in this work.[15–21] The geometry, phonon spectra, and electronic density of states (DOS) are explored to have a better understanding of the experimentally synthesized and theoretically potential Zr-C MXenes.
The rest of this work is organized as follows. The computational methods are described in Sec.
2 Computational Methods
All calculations in this work were performed with the VASP (Vienna Ab-initio Simulation Package) code[22] with projector-augmented plane wave (PAW) potentials[23] and Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional.[24] The wave functions were expanded in a plane-wave basis set with an energy cutoff of 500 eV. A Γ-centered k-point sampling scheme over the Brillouin zone was used for all the calculations. 2D bare and functionalized MXenes were investigated with the vacuum space being larger than 17 Å to avoid any interaction of the MXene sheet with its periodically repeated images along the c axis. A 18 × 18 × 2 k-point mesh was used to optimize the structure of these 2D system. To check the dynamical stability of these 2D system, we calculated the phonon dispersion by using a supercell approach[25] as implemented in the PHONOPY code.[26] In these phonon calculations, a 4 × 4 × 1 supercell and 3 × 3 × 1 k-point mesh were used. The symmetry nonequivalent Zr, Al and C atoms were displaced from their equilibrium positions by an amplitude of 0.02 Å and the atomic forces induced by these small displacements were calculated within VASP. In the calculation of the DOS for pristine and chemical functionalized MXenes, a 42 × 42 × 3 k-point mesh was used.
3 Results and Discussions
3.1 Structural Properties
2 . They are all large negative values, implying that the bare ZrnCn − 1 (n = 2, 3, 4) MXenes tend to be chemically decorated by the oxidizing group. On account of the computed binding energy, the stabilities of the functionalized Zr-C MXenes increase in the sequence of ZrnCn − 1(OH)2 < ZrnCn − 1F2 < ZrnCn − 1O2.
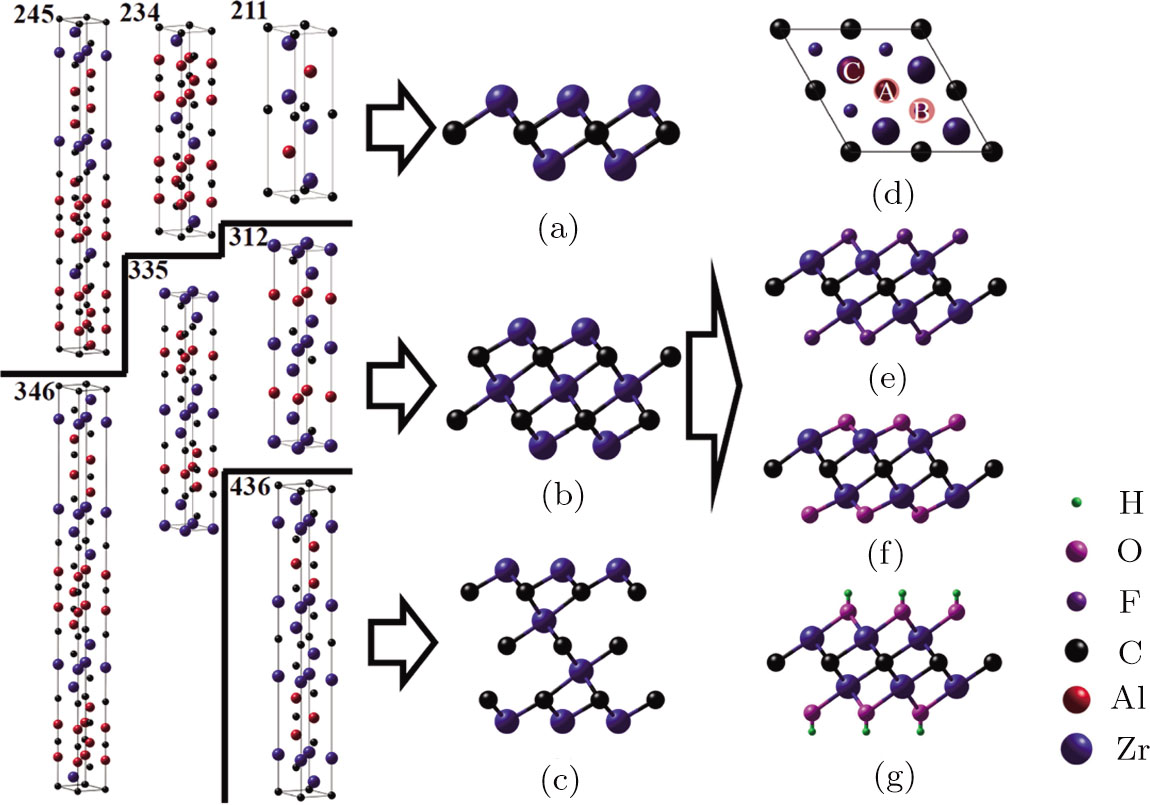
MXene can be prepared from layered nanolaminates by selective etching method. Theoretically there exist three types of Zr-C MXenes ZrnCn − 1 (n = 2, 3 and 4). They can be obtained from the Zr-Al-C nanolaminates (Zr2AlC (211), Zr3AlC2 (312), Zr2Al3C4 (234), Zr3Al3C5 (335), Zr4Al3C6 (436), Zr2Al4C5 (245) and Zr3Al4C6 (346)). The simulated Zr2C, Zr3C2 and Zr4C3 MXene structures as well as their possible precursors in Zr-Al-C system are depicted in Figs.
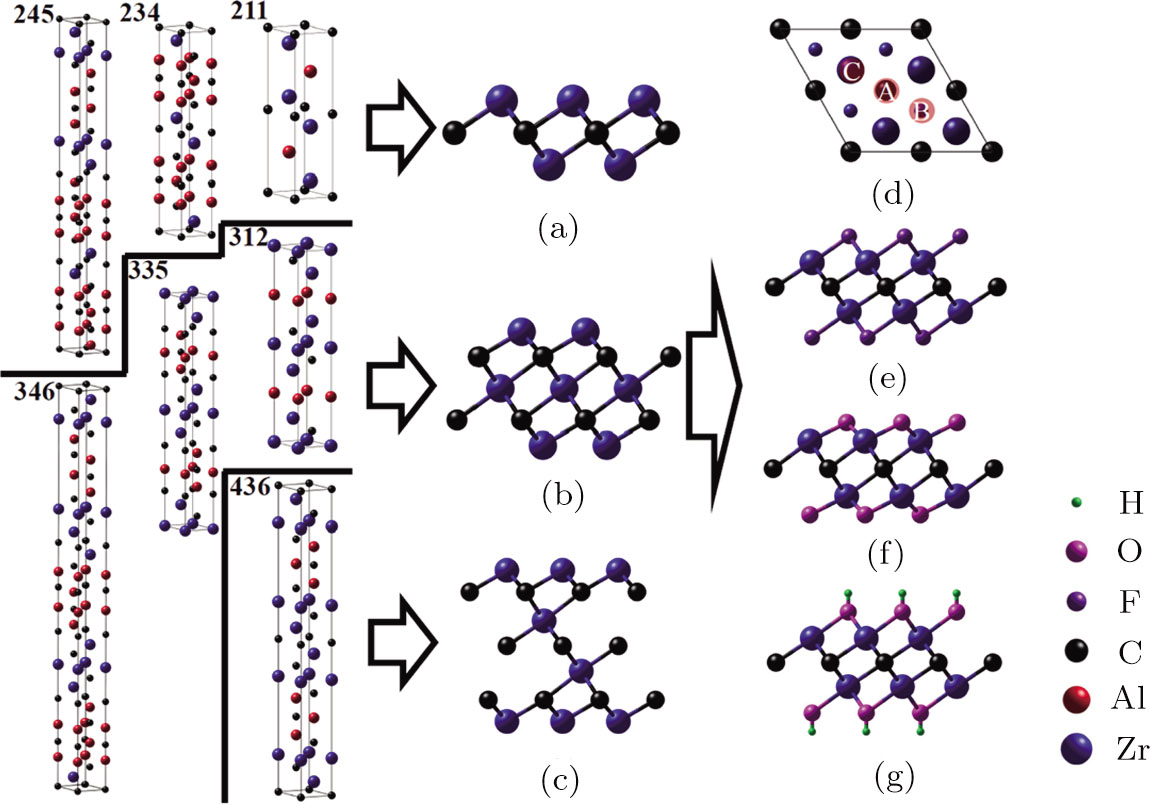 | Fig. 1 (Color online) Schematic illustrations of pristine and functionalized Zr-C MXenes. Side view of pristine Zr-C MXenes as well as their possible precursors in Zr-Al-C system: (a) Zr2C, (b) Zr3C2 and (c) Zr4C3. (d) Top view of Zr2C. The large blue balls are the Zr atoms at the first layer and the small ones are those at the second layer. The symbol A represents the hollow site under which a carbon atom exists, B represents the hollow site under which there is no carbon atom, C is the top site of Zr atom. The rest three graphs are side views of (e) F, (f) O, and (g) OH functionalized Zr2C. The blue, red, black, purple, magenta and green balls indicate the Zr, Al, C, F, O and H atoms, respectively. |
| Table 1
Total energies (eV) of configurations 1-4 for F, O and OH functionalized Zr2C, Zr3C2, and Zr4C3 systems. The most stable functionalization structure is labeled in bold. . |
The optimized lattice parameters of bare3 ZrnCn − 1 (n = 2, 3, 4) and their corresponding F, O and OH functionalized MXenes are shown in Table
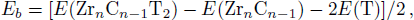
| Table 2 Optimized lattice parameters (Å) and calculated binding energy (eV) of functionalized group in bare ZrnCn − 1 (n = 2, 3, 4), as well as the most stable F, O and OH functionalized configuration. . |
3.2 Vibrational Properties
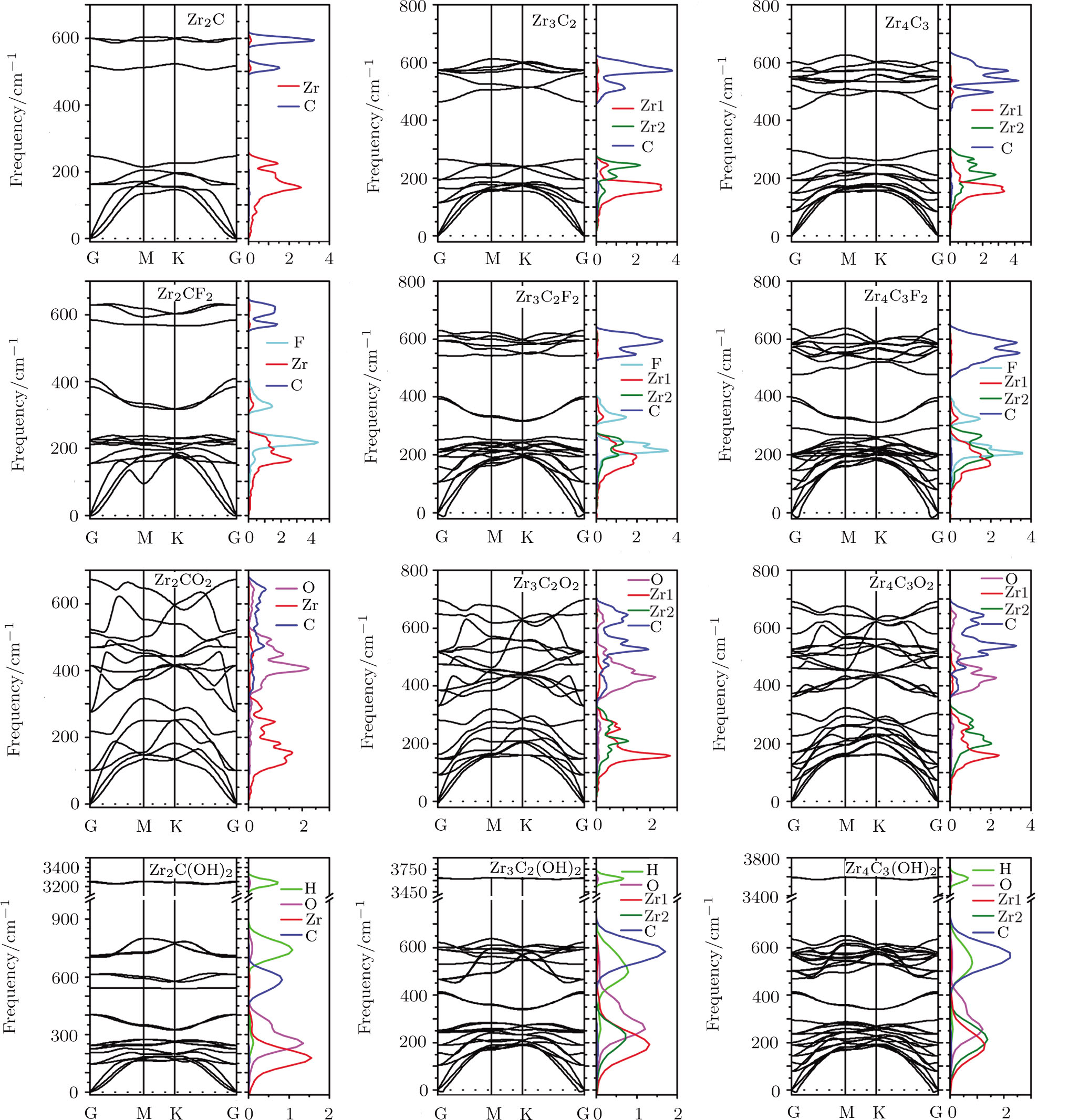
The stabilities of the bare and functionalized Zr-C MXenes are explored by the calculation of their phonon spectra which are depicted in Fig.
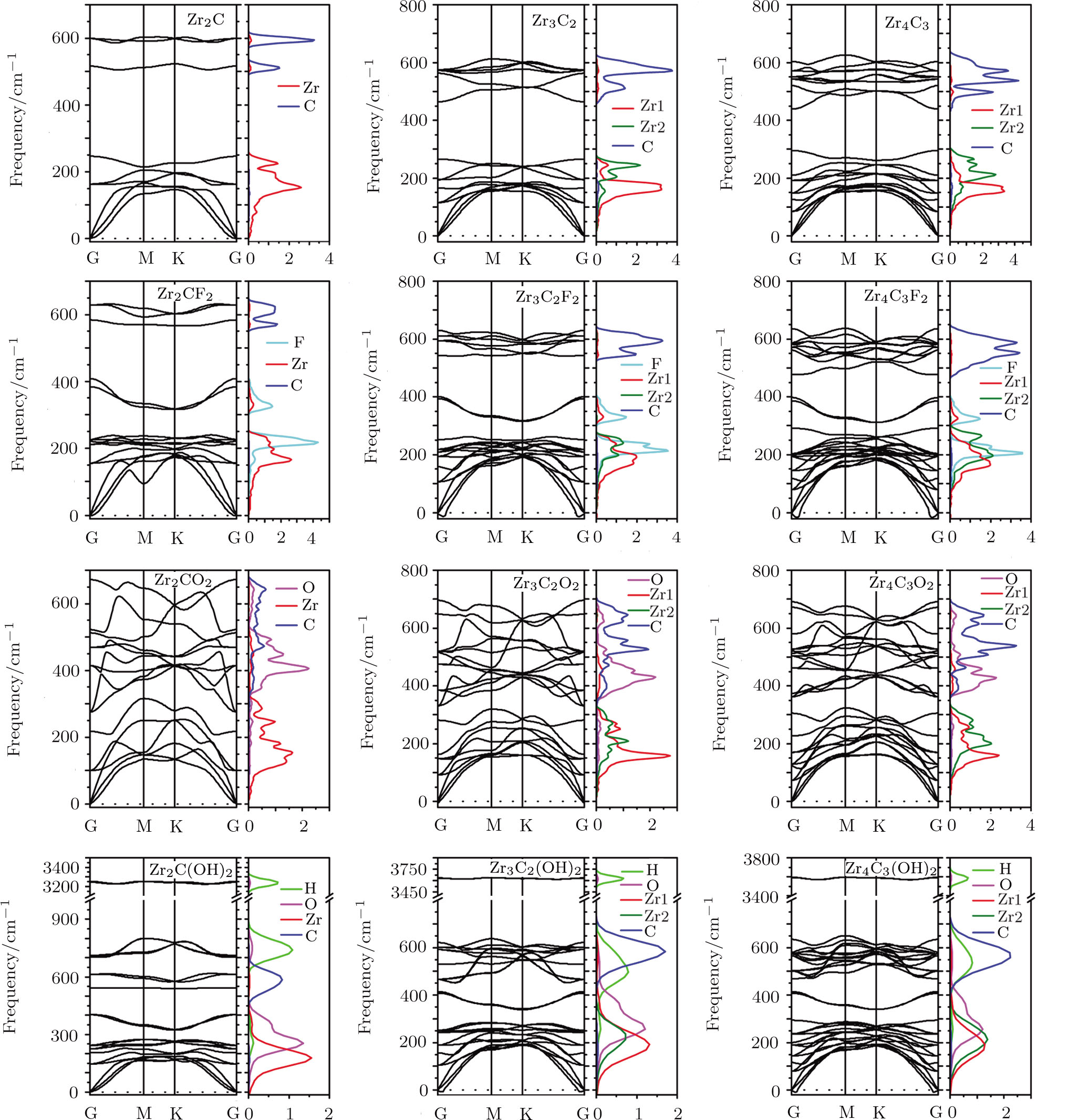 | Fig. 2 (Color online) Phonon dispersion spectra and partial DOS of bare ZrnCn − 1 (n = 2, 3, 4), as well as their most stable F, O, and OH functionalized configuration. Zr represents the superficial Zr layers, and Zr2 is the rest of the Zr layers. |
The phonon partial DOS of Zr-C MXenes are also calculated and depicted in Fig.
3.3 Electronic Properties
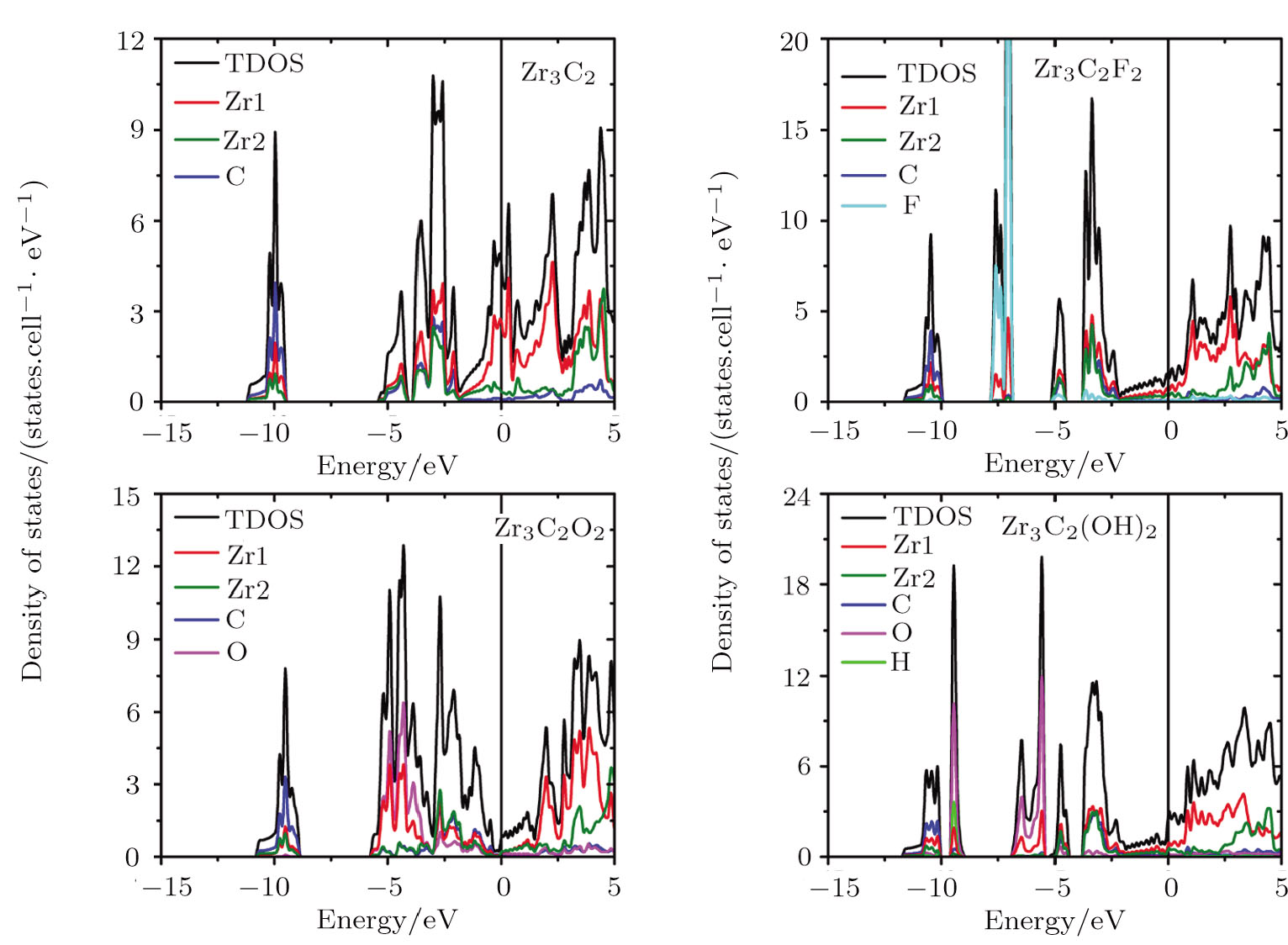
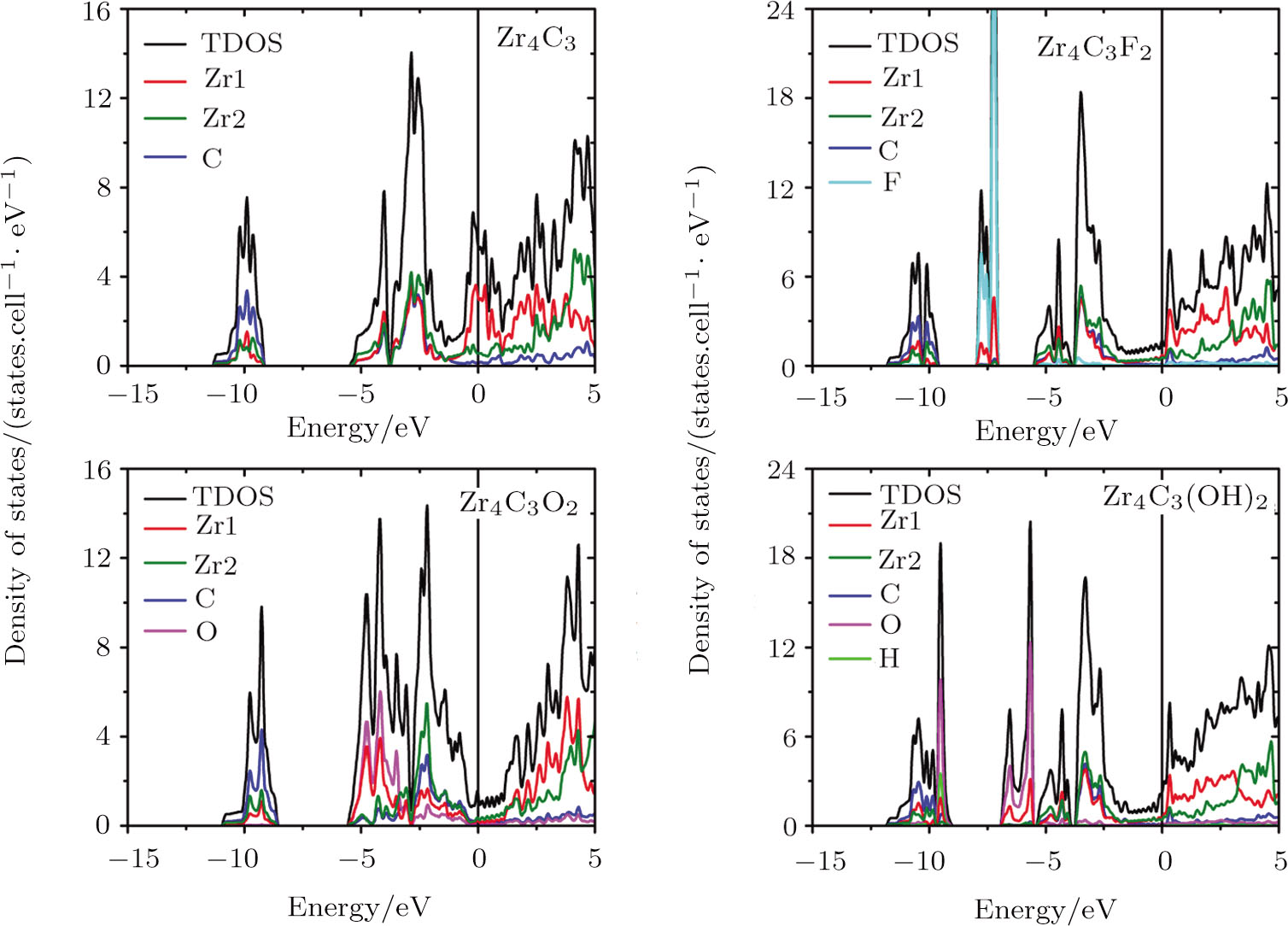
The electronic properties of pristine and surface functionalized Zr-C MXenes have been calculated and presented in Fig.
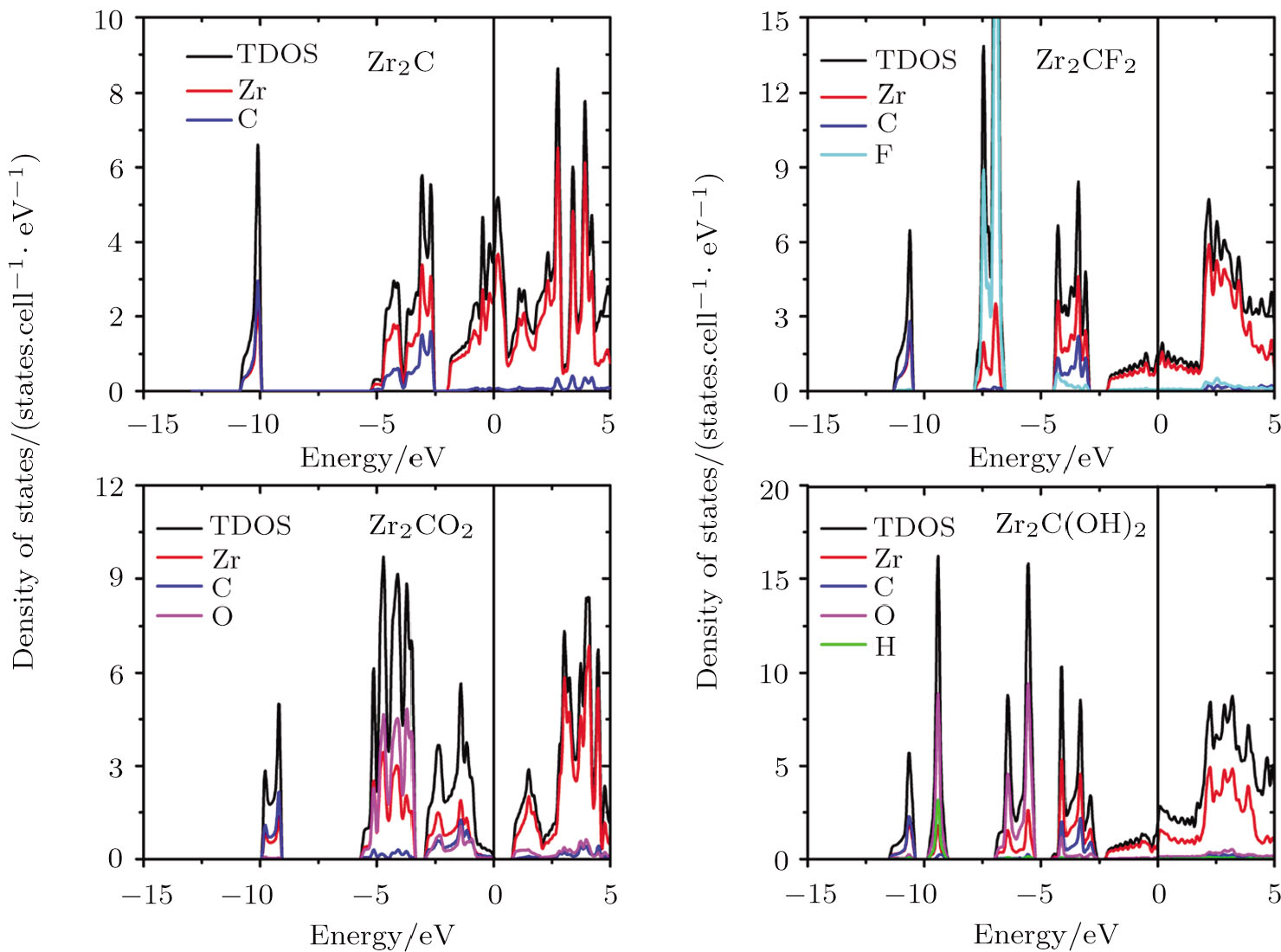 | Fig. 3 (Color online) Total and localized DOS of pristine and surface functionalized Zr2C MXenes. The vertical line represents the Fermi level. |
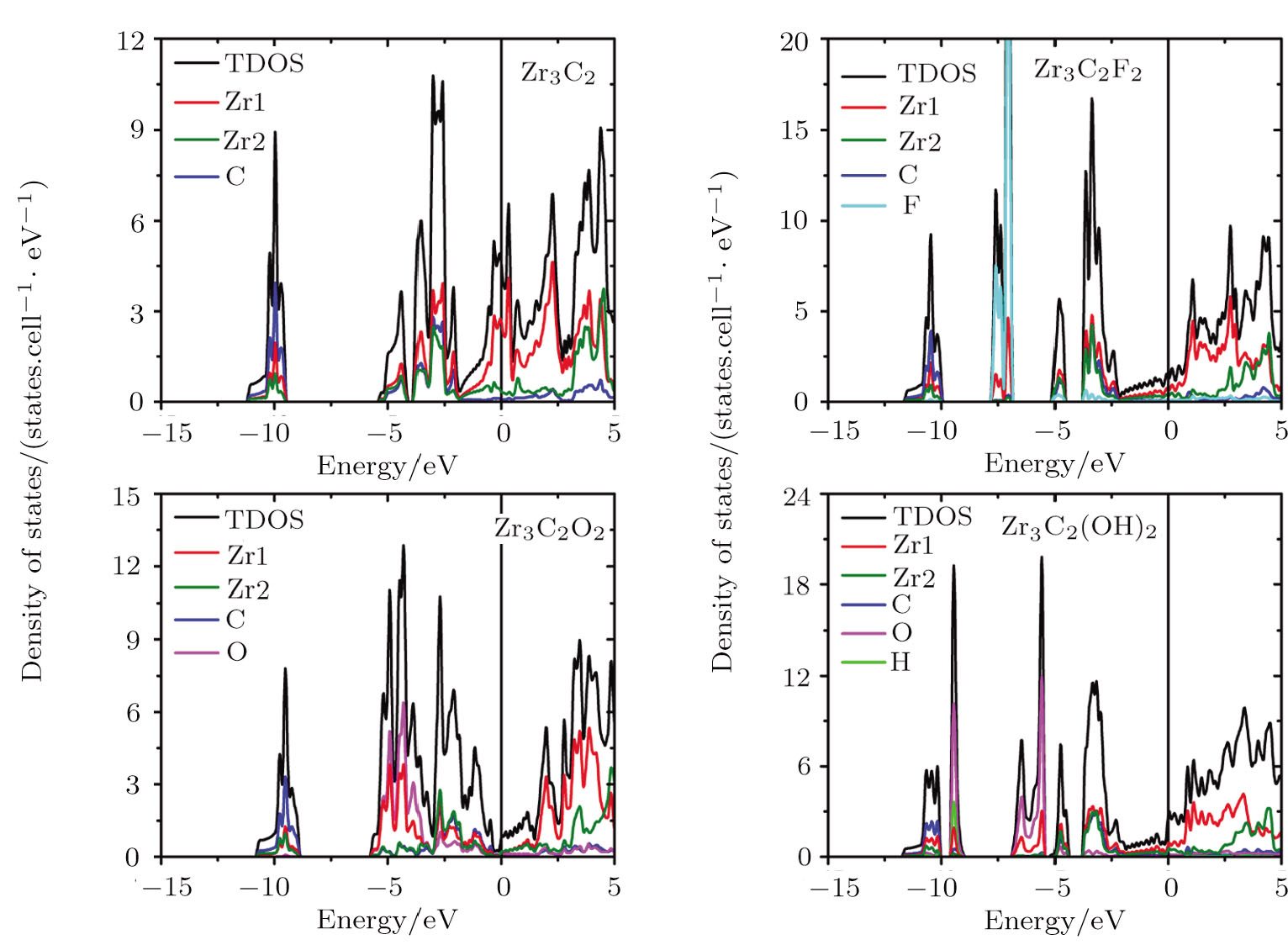 | Fig. 4 (Color online) Total and localized DOS of pristine and surface functionalized Zr3C2 MXenes. Zr1 represents the superficial Zr layers, and Zr2 is the middle Zr layer. The vertical line represents the Fermi level. |
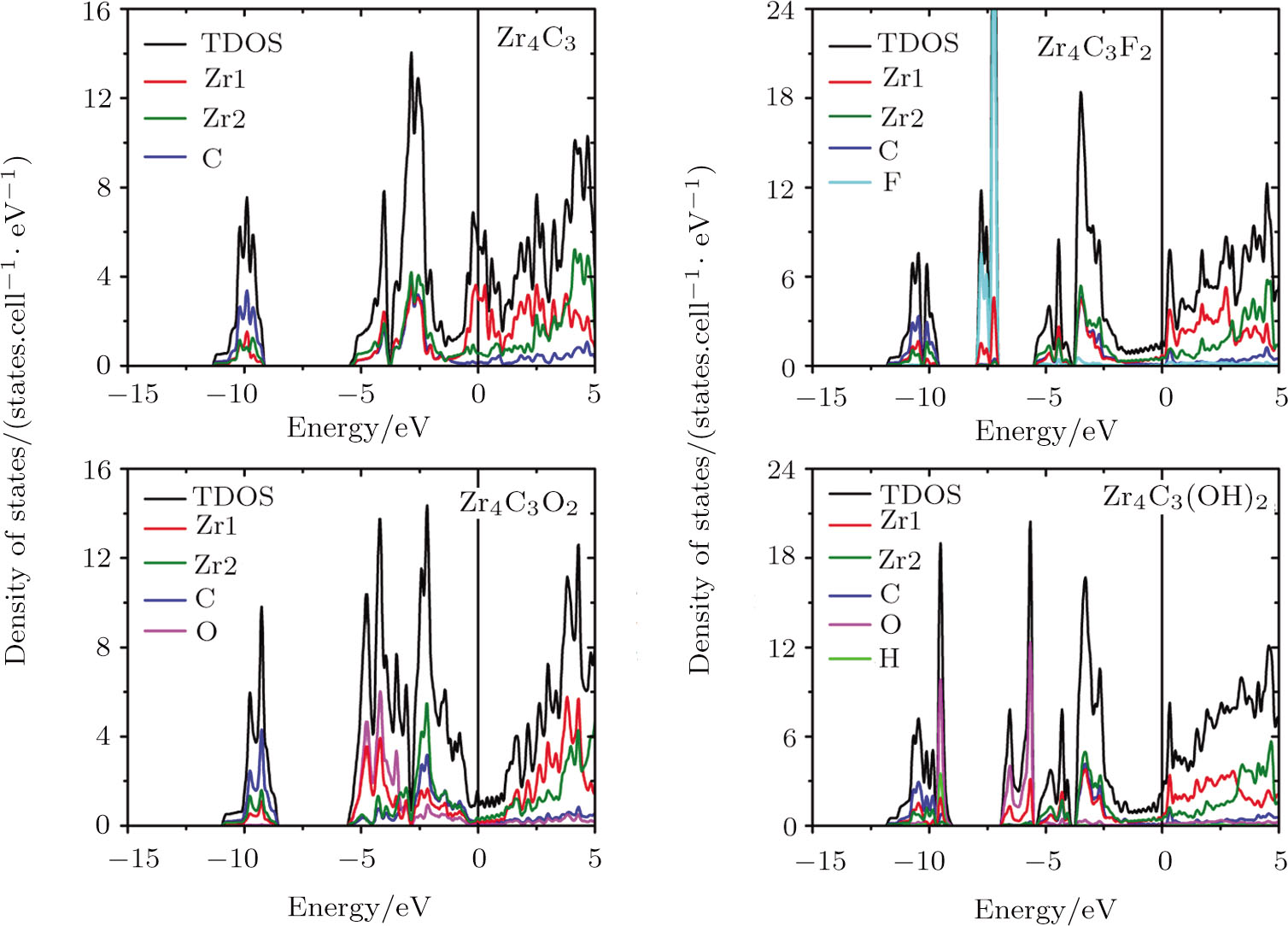 | Fig. 5 (Color online) Total and localized DOS of pristine and surface functionalized Zr4C3 MXenes. Zr1 represents the superficial Zr layers, and Zr2 is the rest of the Zr layers. The vertical line represents the Fermi level. |
When ZrnCn − 1 (n = 2, 3, and 4) MXenes are functionalized by F, O, and OH, new hybridization states appear and the DOS of MXenes at the Fermi level are reduced. The adsorbed F group hybridizes with the Zr atom at about −7.5 eV in all kinds of Zr-C MXenes and obtains one electron from the pristine ZrnCn − 1 (n = 2, 3, and 4). The new hybridization state between hydrogen and oxygen atoms of the hydroxyl group locates around −9.5 eV in OH terminated Zr-C MXenes. Other additional bands in hydroxyl functionalized Zr-C MXenes are at about −6 eV and they are contributed by the interaction of the O atoms and the superficial Zr atoms. The functionalized O atom gives rise to large DOS in ZrnCn − 1O2 (n = 2, 3, and 4) at about −4.4 eV and acquires two electrons from the pristine ZrnCn − 1 (n = 2, 3, and 4). The Fermi energy of the O terminated ZrnCn − 1 (n = 2, 3, and 4) MXenes shifts downward to the right edge of the hybridization state of C atom and Zr atom. In Zr2CO2, the Fermi level situates just at the band gap of the empty Ti band and the hybridized band of Zr, C and O.
In Zr3C2 and Zr4C3, the superficial Zr layers hybridize with the functionalized group in the low energy region, while the rest of the Zr atoms interact with the C atoms in the relatively high energy region. As the n value of (ZrnCn − 1) and (ZrnCn − 1)T2 decreases, the thicknesses of Zr slabs gradually decline, which lowers the DOS near the Fermi level. Especially for (ZrnCn − 1)O2, Zr4C3O2 is still a metal, Zr3C2O2 is a semimetal, while Zr2CO2 becomes a semiconductor with a band gap of 0.8 eV.
4 Conclusions
The fundamental properties of Zr-C MXenes are important to their potential applications. In this work, a systematic first-principles study on the structural, vibrational, and electronic properties of Zr-C MXenes have been conducted. The most stable configurations for Zr-C MXene are the ones that the terminal groups F, O, and OH locate on the common hollow site of the superficial Zr layer and its adjacent C layer. The stabilities of the functionalized Zr-C MXenes increase in the sequence of ZrnCn − 1(OH)2 < ZrnCn − 1F2 < ZrnCn − 1O2 according to the species of the terminal group. Among all the investigated MXenes, F and OH-terminated Zr3C2 and Zr4C3 have small imaginary acoustic phonon branches around Γ point while the others have no negative phonon modes. The pristine MXenes (Zr2C, Zr3C2 and Zr4C3) are all metallic with large DOS contributed by the Zr atom at the Fermi energy. When functionalized by F, O and OH, new hybridization states appear and the DOS at the Fermi level are reduced. Especially for ZrnCn − 1O2, Zr4C3O2 is still a metal, Zr3C2O2 is a semimetal, while Zr2CO2 becomes a semiconductor.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] |