Application of Connection in Molecular Dynamics
Cite this Article
Sun Xin. Application of Connection in Molecular Dynamics. Communications in Theoretical Physics, 2018, 69(3): 308 
Permissions
Application of Connection in Molecular Dynamics
† Corresponding author. E-mail:
Abstract
Abstract
The evolution of electronic states in molecule has two origins: dynamical one produced by Schrödinger equation and kinematical one caused by base transformation due to nuclear motion. In current theories, the former gets analytic expression; the latter depends on heavy numerical calculation, which contains uncertainty. By using connection of fiber bundles, this paper establishes an analytic formula for the latter, and the numerical work is simplified. It shows the mathematical structure of molecule is fiber bundle.
1 Introductionx , R ) is the interaction between electrons and nuclei. In H(x |R ), R are parameters, the eigen-equation of H(x |R ) isx | R ) constitute an orthogonal complete set, which can serve as the base vectors of a Hilbert space.
In molecule, since nuclei are much heavier than electrons, they are considered as classical particle, this is semi-quantum molecular dynamics. The electronic states in molecule are time-dependent superpositions of all the eigenstates, and the electron distribution is changing with the time, this is dynamical evolution. At the same time, the nuclear motion makes the base vectors of the eigenstates continuously varying, and it indirectly induces additional evolution, it is kinematical evolution.
Recent years, the semi-quantum molecular dynamics, especially the Ehrenfest dynamics, have got further progress,[1–4] where the dynamical evolution can be expressed analytically, and its calculation is transparent. However, the kinematical one depends on complicated numerical calculation,[5–7] which causes some confusions. It is needed to establish an analytic formula to express the kinematical evolution, then all the results become definite. This paper will provide one approach based on the connection in the geometry to derive such an expression.
A molecule consists of N nuclei and M electrons, the coordinates of nuclei are {
The electronic Hamiltonian is
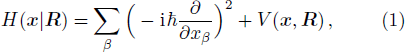
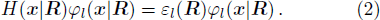
2 FormulationR and corresponding complete set of φm(x | R ), the electronic state ψk(x , t) (k is the number of the state) can be expanded by φm(x | R )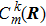
R ) is the interaction among nuclei.R )|∇ H(R )|φn(R )⟩ is easy to get by numerical calculation, it is the key factor to solve the Newtonian Eq. (7 ). Later, it can be seen that the gradient matrix will play an important part also in solving Schrödinger equation.R moves ΔR , which is determined by the Newtonian Eq. (7 ).4 ) and Eq. (9 ) into Eq. (8 ),x | R′ ) | × Eq. (10 )
The time-dependent Schrödinger equation is
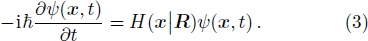
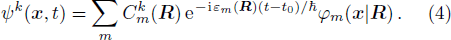
Actually Eq. (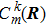
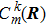
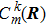
At the beginning t = t0,

Nuclei obey the Newtonian equation
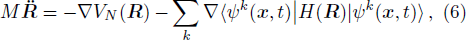
By using the generalized Hellmann-Feynman Theorem,[8] Eq. (
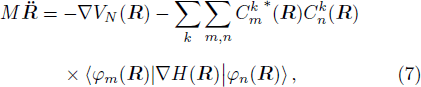
By solving the combined equations of Schrödinger Eq. (
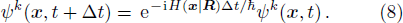
For each step, during the interval Δt, the configuration is
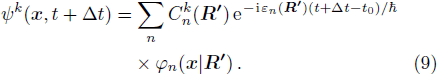
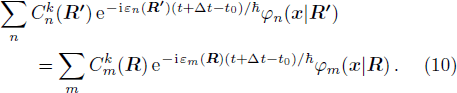
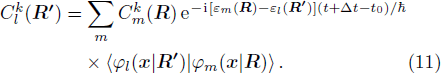
From Eq. (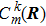
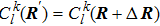
Thus, the evolution of the electronic state is demonstrated.
In each step, the starting time t can be taken as t0. Then Eq. (
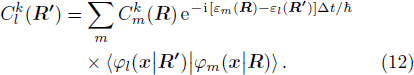
This equation shows that the evolution of the electronic state 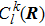
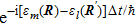
The rest problem is how to calculate the matrix ⟨φl(
3 Connection of Fiber BundleR′ , R ) is the transformation matrix between the base vectors φn(x |R ) and φn(x |R′ ).R′ , R ) satisfies the multiplier rule, so Dnl(R′ , R ) is a representation of the deformation group.R ′,R ) in the component Rα is18 ) into Eq. (17 ),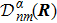
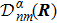
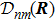
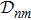
R )|∇ H(R )| φn(R )〉, which has been got in Newtonian Eq. (7 ).22 ) into Eq. (12 ), we get23 ), the last term 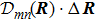
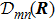
20 )) of the fiber bundle, and original heavy numerical calculation is avoided. If neglecting the connection the electronic state would not be able to spread over different eigenfunctions, that is the BOA.
The change from configuration
In base space
During the deformation from
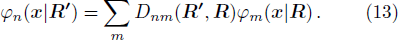
From Eq. (
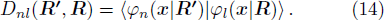
Very close to
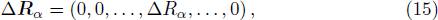
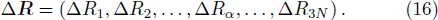
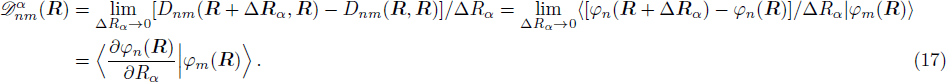
In geometry, this rate 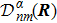
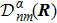
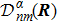
By the way, in algebra, 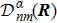
By using the perturbation theory, we can get
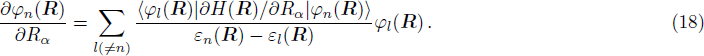
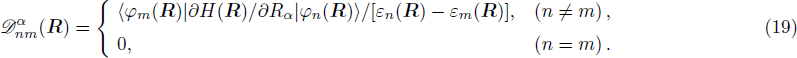
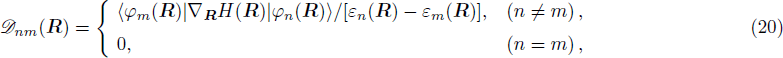
By using 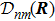
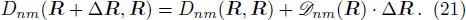
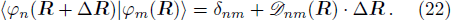
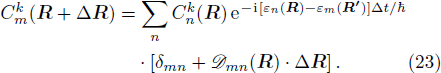
Finally, by solving combined equations (
Polymers are chain-like molecules. Since they are one-dimensional systems, the polymers have prominent self-trapping, which is a typical deformation effect. The self-trapping produces many novel phenomena in polymers, such as photoinduced spin-flipping of charge carries and charge-flipping of spin carries. The Ehrenfest dynamics has been used to study these processes.[6–7] But these works have a problem, that their numerical calculations are very heavy, even getting some uncertainty. The connection of fibre bundle can be used to reduce the numerical calculation and make results transparent.
4 Conclusion
Molecule is a physical model of the fiber bundle, which can be used to build analytic expression for molecular dynamics and simplify numerical calculation.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] |